
Тхана Прасонгсин / Гетти Имагес
Кључне Такеаваис
- Постоји само један третман који је одобрила ФДА за болест српастих ћелија, али за њега је потребан донор браће и сестара.
- Користећи ЦРИСПР-ЦАС9 технологију, истраживачи су били успешни у циљању генетског прекидача који је искључио производњу феталног облика хемоглобина.
- ЦРИСПР-ЦАС9 омогућио је пацијентима са болестима српастих ћелија и бета-таласемијом да буду сами донатори за трансплантацију коштане сржи. Ово може учинити третмане широко доступним.
Уређивање гена преокренуло је многа подручја науке, од стварања хране без пестицида до покушаја враћања вуненог мамута. Користећи технологију названу ЦРИСПР-ЦАС9, која се обично назива ЦРИСПР, научници сада покушавају да поправе генетске грешке које узрокују болест.
Јануарска студија објављена уТхе Нев Енгланд Јоурнал оф Медицинеоткрио је да ЦРИСПР може створити нове третмане за лечење поремећаја крви као што је болест српастих ћелија.
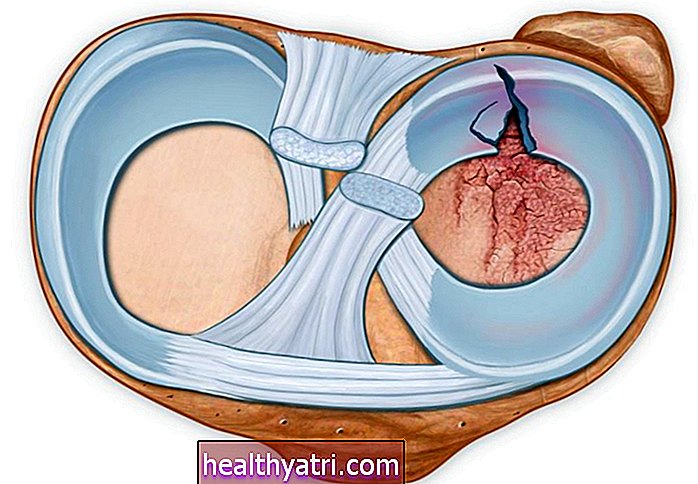
Пацијенти којима је дијагностикована болест српастих ћелија имају мутацију гена за хемоглобин - протеин богат гвожђем у црвеним крвним зрнцима. Мутација узрокује абнормалне крвне ћелије у облику слова Ц. Тешко преносе кисеоник у друге делове тела. Његова тврда и лепљива особина такође зачепљује проток крви, што повећава ризик од инфекција.
Болест српастих ћелија је наследни поремећај крви који погађа око 100 000 Американаца годишње, каже Алекис А. Тхомпсон, МД, МПХ, бивши председник Америчког друштва за хематологију и дечији хематолог на Медицинском факултету у Феинбергу на Универзитету Северозапад, за Веривелл. Иако Томпсон, који није био укључен у студију, каже да се деца при рођењу чине релативно нормалним, тек од 6 до 12 месеци деца почињу да развијају проблеме.
„У млађој старосној групи пацијенти који имају бол, јаку температуру или инфекцију захтевају хоспитализацију, примају врло јаке лекове и пропуштају школовање“, каже Томпсон. „Како прелазе у одраслу доб, изазови су завршавали образовање, факултет или задржавање посла “. Користећи ЦРИСПР, истраживачи покушавају да промене неке од ових исхода.
Шта ово значи за вас
Болест српастих ћелија преноси се на дете када оба родитеља имају својство српастих ћелија. Ако нисте сигурни у свој статус превозника, неопходно је да вас прегледа здравствени радник. Ако имате болест српастих ћелија, нови третмани који користе ЦРИСПР технологију можда ће вам бити доступни у будућности.
Генетска стратегија поново покреће производњу хемоглобина
Студија је пратила једног пацијента са болешћу српастих ћелија и једног пацијента са бета-таласемијом, поремећајем крви који смањује производњу хемоглобина.
Обојици пацијената биле су потребне матичне ћелије крви, али студија је тежила да користи њихове ћелије, а не ћелије брата или сестре. Када су пацијенту узете матичне ћелије крви, истраживачи су користили ЦРИСПР, који делује као молекуларна маказа, и молекул РНК са једним водичем, ЦАС9, за лоцирање специфичног гена названог БЦЛ11А.
У овој студији истраживачи су пресекли БЦЛ11А јер делује као генетски прекидач који искључује ген који ствара фетални облик хемоглобина. Поновним укључивањем, научници су реактивирали производњу феталног хемоглобина, који је заменио недостајући или неисправни хемоглобин у црвеним крвним зрнцима оба пацијента. Све преостале болесне ћелије елиминисане су хемотерапијом.
Нивои хемоглобина остали су стабилни месеци након лечења
Шест и 12 месеци након поступка, оба пацијента су подвргнута аспирацијама коштане сржи како би измерили број црвених крвних зрнаца присутних у њиховом узорку кости.
Први пацијент била је 19-годишња жена са дијагнозом бета-таласемије. Четири месеца након последње трансплантације коштане сржи са генетски уређеним матичним ћелијама, ниво хемоглобина се стабилизовао и остао стабилан током последње посете. Иако је у почетку имала озбиљне нежељене ефекте лечења (упала плућа и болести јетре), они су се повукли након неколико недеља.
Други пацијент је била 33-годишња жена са болешћу српастих ћелија. Петнаест месеци након поступка, ниво њеног феталног хемоглобина порастао је са 9,1% на 43,2%. Њени мутирани ниво хемоглобина од српастих ћелија смањио се са 74,1% на 52,3%. Док је имала три тешка нежељена ефекта (сепсу, холелитијазу и болове у стомаку), они су решени лечењем.
Једна од главних предности овог приступа, у поређењу са традиционалним облицима лечења ових поремећаја крви, је употреба ћелија пацијента без потребе за даваоцем.
„Ћелијама истог пацијента може се манипулисати и могу се трансплантирати без ризика од одбацивања или изазивања имунолошких реакција даваоца (болест трансплантата против домаћина)“, Дамиано Ронделли, др. Мед., Професор хематологије Мицхаел Реесе на Универзитету из Илиноиса на Медицинском факултету у Чикагу, наводи се у изјави.
Од објављивања, истраживачи су проширили свој рад на још осам пацијената - шест са бета-таласемијом и три са болешћу српастих ћелија. Њихови тренутни резултати су у складу са прва два пацијента у студији.
Тренутни третман српастих ћелијских болести
Тренутно лечење болести српастих ћелија које је одобрила ФДА је трансплантација коштане сржи. Међутим, овај поступак захтева да пацијент има брата и сестре чије се ткиво савршено подудара са њиховим ткивом.
Томпсон каже да је главни изазов за лечење тај што сваки четврти браћа и сестре нису исте врсте ткива. Чак и ако се изврши трансплантација коштане сржи, постоје и озбиљни нежељени ефекти на поступак, укључујући отказивање графта, болест калем против домаћина и смрт.
Ако се трансплантација коштане сржи избаци из слике, алтернативни начин лечења је хаплоидно идентична трансплантација. „Постигнут је успех са хаплоидним идентичним трансплантацијама где се тип ткива делимично подудара, али трансплантација се изводи на сасвим другачији начин да би се постигло усађивање уз ваше компликације“, каже Томпсон. Међутим, она каже да се само мањи број пацијената квалификује за овај третман.
Због ограничења и ограничења за болест српастих ћелија, Томпсон каже да је било неких расправа о томе да пацијенти буду сами себи донатори. У овој тренутној студији аутори на уређивање гена гледају као на потенцијални пут за ову врсту лечења.
Како генетски третмани могу помоћи
Свако може наследити болест српастих ћелија, али то је посебно често код:
- Људи афричког порекла, укључујући Афроамериканце
- Латиноамериканци из Централне и Јужне Америке
- Људи блискоисточног, азијског, индијског и медитеранског порекла
У САД-у се сва деца рођена у земљи прегледају на болест српастих ћелија, што даје довољно могућности за рано лечење. Али неколико сценарија представља изазов за дијагнозу сваког случаја. Томпсон каже да породице које су имигрирале у САД могу имати старију децу која нису прегледана заједно са родитељима који нису свесни свог статуса носиоца док не добију дете које има такво стање.
Упркос несавршености скрининга, индустријске земље су побољшале прогнозу болести српастих ћелија. „Данас дете рођено данас у Сједињеним Државама има 95% шансе да преживи у одраслој доби, а исто важи и за друге сналажљиве земље попут Уједињеног Краљевства“, каже Томпсон.
Из глобалне перспективе, међутим, Томпсон каже да земље са ниским и средњим приходима можда неће понудити исте третмане који су тренутно доступни људима у земљама попут САД-а.Каже да преко половине деце са болестима српастих ћелија у подсахарској Африци неће преживети свој пети рођендан.
На основу резултата студије, уређивање гена могло би да помогне да третмани за болести српастих ћелија постану шире доступни.
„Нада се да ће овај третман бити доступан и приступачан у многим земљама са ниским средњим приходима, Блиском Истоку, Африци и Индији и да ће имати важан утицај у животу многих људи на овим просторима“, рекао је Ронделли. У





















.jpg)